Electrophoretic
Analysis of DNA **Practice
Plasmid.exe
-- It will be on the Quiz**
Cloning
genes
PCR
Gene
sequence analysis
Complementarity
and Hybridization
Northern,
Southern, and Western blots
DNA
Microarrays
Transgenics
What
is gene technology? All gene
manipulation
is based on microbial genetics--ways of doing in the test tube what
bacteria
and viruses do naturally. Several of this week's gene manipulations are
exemplified by the article on overexpressed
angiopoietin in a transgenic mouse.
Gel
Electrophoresis Images and text based on MIT
Hypertextbook
This technique
separates
molecules on the basis of their size.
-
Cast slab
of gel material,
usually agarose or polyacrylamide. The gel is a matrix of
polymers
forming sub-microscopic pores.
-
The size
of the pores
can be controlled by varying the chemical composition of the gel.
The gel is set
up for
electrophoresis in a tank holding pH buffer. Electrodes
apply
an electric field:
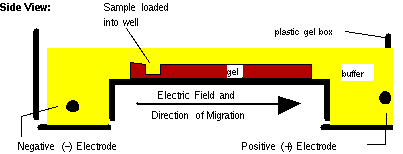
MIT
hypertextbook
The
molecules to separate
(DNA RNA) carry a net negative charge (why?)
so
they move along the electric field toward the positive
cathode. (To separate proteins, a detergent would be
included which coats the protein with negative charge.)
The larger
molecules
are held up as they try to pass through the pores of the gel, while the
smaller molecules are impeded less and move faster. This results
in separation by size, with the larger
molecules
nearer the well and the smaller molecules farther away.
Note that
this separates
on the basis of size (volume in solution),
which is not necessarily molecular weight.
For example:
-
Two DNA
molecules of the
same molecular weight will run differently if one is supercoiled,
because
the supercoils constrain the shape to be smaller.
-
Two RNA
molecules of the
same molecular weight will run differently if one has much
intramolecular
base pairing, making it "smaller."
Aside from the
above exceptions,
the distance migrated is roughly proportional to
the log of the inverse of the molecular weight (the log of
1/MW).
Gels are normally depicted as running vertically, with the wells at the
top and the direction of migration downwards. This leaves the large
molecules
at the top and the smaller molecules at the bottom. Molecular weights
are
measured with different units for DNA, RNA, and protein:
-
DNA:
Molecular weight is measured in base-pairs, or bp, and commonly in
kilobase-pairs
(1000bp), or kbp.
-
RNA:
Molecular weight is measured in nucleotides, or nt, and commonly in
kilonucleotides
(1000nt), or knt. [Sometimes, bases, or b and kb are used.]
-
Protein:
Molecular weight is measured in Daltons (grams per mole), or Da, and
commonly
in kiloDaltons (1000Da), or kDa.
Molecular
weight standards
run in one well of the gel are used to calibrate the molecular weights
of sample molecules. Below is a gel stained with a dye: a colored
molecule which binds to a specific class of macromolecules in a
sequence-independent
manner (probes bind in a sequence-dependent manner).
Sample
1 contains only one size class of macromolecule - it could be a
plasmid, a pure mRNA transcript, or a purified protein. In this case,
you
would not have to use a probe to detect the molecule of interest since
there is only one type of molecule present. Blotting is usually
necessary
for samples that are not complex mixtures. By interpolation, its
molecular
weight is roughly 3.
Sample
2 is what a sample of total DNA cut with a restriction enzyme,
total
cellular RNA, or total cellular protein would look like in a gel
stained
with a sequence-independent stain. There are so many bands that it is
impossible
to find the one we are interested in. Without a probe (which acts like
a sequence-dependent stain) we cannot get very much information from a
sample like this.
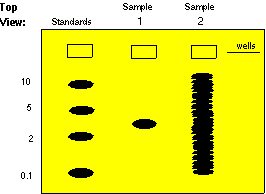
MIT
hypertextbook
Different stains
are used for different classes of macromolecules. DNA and RNA are
generally stained with ethidium bromide (EtBr),
an
intercalating agent. The DNA-EtBr complex fluoresces under
UV light. Protein is stained with Coomassie Blue or Silver
Stain.
Cloning
genes
In nature, DNA molecules
recombine
for various functions -- even DNA between different species. But
twenty years ago, despite the work of Barbara McClintock and others,
the
extent of this recombination was not appreciated. DNA was still
thought
to be the "master molecule," not to be violated by "unnatural"
manipulation.
When scientists began to manipulate DNA in the test tube, many
scientists
feared that disastrous monsters would result, with unspecified dangers
to people. In 1977 scientists at the Asilomar Conference proposed
sweeping regulation on so-called "recombinant DNA," technologies which
recombine DNA from different species in the test tube.
Since then, the dangers
have appeared
to be little more than those of "natural" genetic mixing.
But
we remain concerned about issues such as:
-
Engineering food crops
to resist
pesticides. The pesticide resistance genes can escape into
natural
populations of weeds.
-
Engineering a human
symbiont microbe,
such as E. coli, to produce a deadly toxin such as
botulin.
In theory this could be done, although it's not clear where such an
organism
would live, or how well it could "compete" with natural flora.
-
Societal dilemmas of
human cloning.
How far shall we use reproductive technology to shape future humans?
Techniques
of Recombinant
DNA
How do we manipulate these
natural
processes for biotechnology; for instance, to make a bacterium that
produces
large quantities of insulin?
One approach would be to
cut the
appropriate gene from human DNA and paste, or splice, it into a vector
such as a plasmid or phage DNA. Our "scissors" are the class of
enzymes
called restriction endonucleases
Restriction
Endonucleases
An "endonuclease" is an
enzyme
that cuts duplex DNA in the middle, not at an end (for
exonuclease).
Different species of bacteria have evolved different restriction
endonucleases,
each to cut foreign DNA that gets into their cells by mistake. To
be cut, the DNA has to lack their own pattern of protective
methylation.
There are well over a hundred restriction enzymes, each cutting in a
very
precise way a specific base sequence of the DNA molecule.
A restriction
endonuclease cuts
DNA only at a specific site, usually containing 4-6 base pairs.
The
enzyme has to cut the DNA backbone twice, recognizing the same type of
site; therefore, the site "reads" the same way backwards as forwards--a
palindrome.
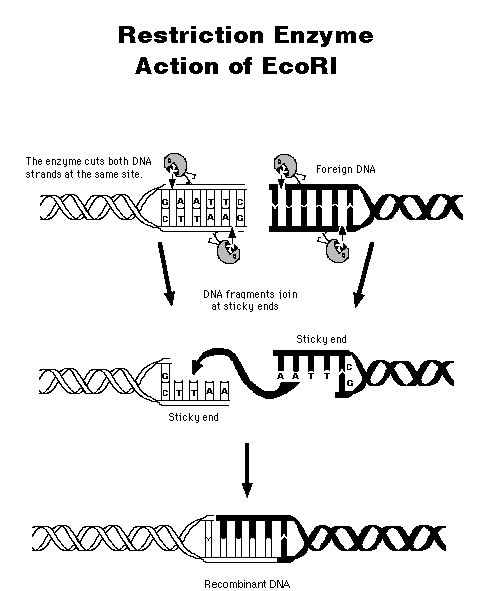
This
"sticky ends"
from two different DNA molecules can hybridize together; then the nicks
are sealed using ligase.
(Where does ligase come from? What is its natural function?) The
result is recombinant DNA.
When
this recombinant vector is inserted into E. coli, the cell will be able
to process the instructions to assemble the amino acids for insulin
production.
More importantly, the new instructions are passed along to the next
generation
of E. coli cells in the process known as gene
cloning.
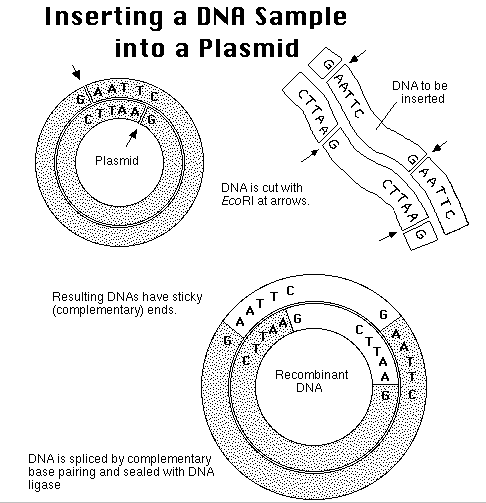
Restriction
site
Analysis
How can we use restriction
sites
to analyze the plasmid products of ligation, and tell whether we in
fact
have ligated the correct molecule:
Problem:
Suggest several "incorrect" ways the plasmid could recombine.
More
Problems:
Use
the
PLASMID
Program. You MUST practice restriction analysis with this
program; it will be on the quiz and/or the test.
How do we get the
recombinant
molecule into a bacterial cell? Usually by transformation
(for a plasmid) or by in vitro packaging
into a phage head coat (for a phage vector such as lambda phage).
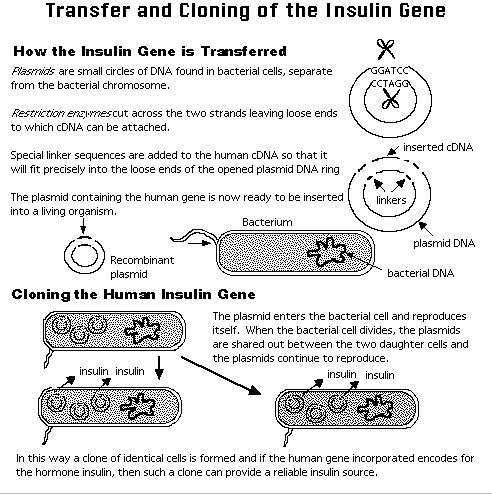
The above is a highly
simplified
description of recombinant DNA technology.
How would we actually
locate
the appropriately cloned gene? There are many different ways,
depending
on the specific case. Here is one example, in which a partial
sequence
of the protein enables us to reverse the code and
determine an approximate DNA sequence to use for a radiolabeled
probe. The DNA probe will hybridize
to clones containing the correct DNA, even if it is just one piece cut
out of an entire genome.
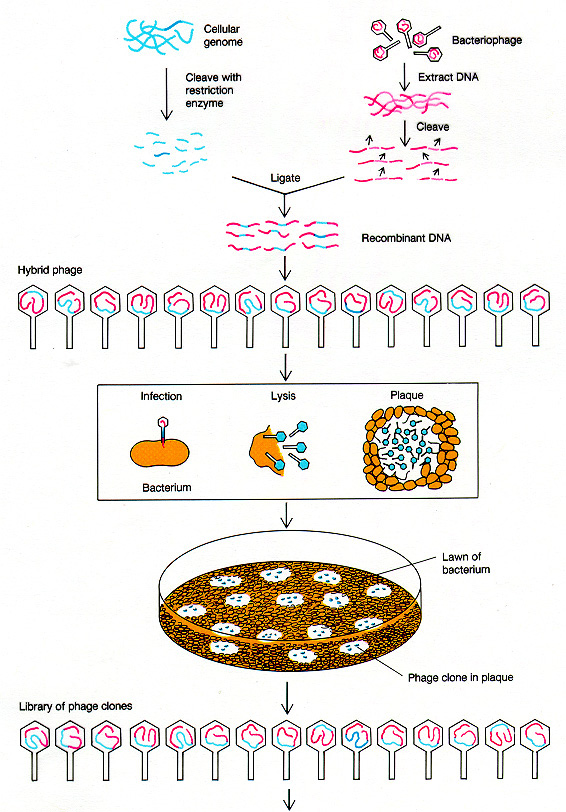
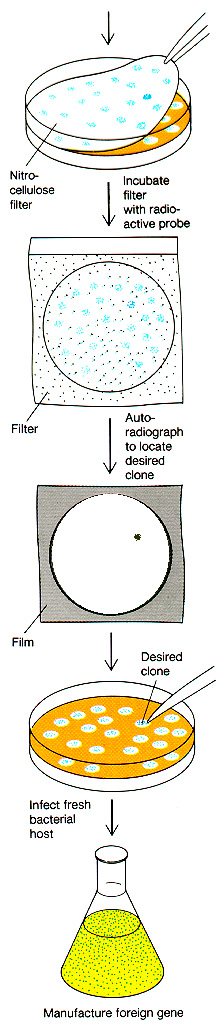
Griffiths
et al, W. H. Freeman & Co., current edition
The radioactive
probe is made by determining a short
segment
of the protein sequence, then "back translating" to the possible DNA
sequences.
Short DNA sequences are synthesized to match the protein
sequence.
Then these DNA oligomers (known as "oligos") are radiolabeled, and
applied
to the blotted clones. They should hybridize only to clones
containing
sequence encoding the desired protein.
Reverse
transcription:
cDNA Cloning
Suppose we need to clone a
gene
containing lots of introns. What will
happen
when the bacterium tries to express it?
To
overcome
this problem, we can start with mRNA
isolated from tissues that produce the desired protein. We
then use reverse transcriptase enzyme
(produced by a retrovirus related to HIV) to reverse transcribe the
mRNA
into a DNA molecule that now is free of introns. Now we can
ligate
"sticky ends" onto the cDNA
and recombine it into a phage or plasmid vector.
Problem:
Know the differences between genomic cloning and cDNA cloning.
Explain
the relative ADVANTAGES and DISADVANTAGES of each technique--depending
on the aim of your research.
Gene Therapy
Transgenic Plants
PCR:
Summary of technique
In
PCR,
a heat-stable DNA polymerase is used, most commonly Taq
Polymerase from the thermophilic microbe Thermus
aquaticus. Thomas Brock discovered T. aquaticus
from a hot spring at Yellowstone National Park.
 Prismatic
Pool, Yellowstone Prismatic
Pool, Yellowstone
More
recently,
an even more heat-resistant polymerase has been developed from a
hyperthermophilic
microbe growing at 110 degrees C in hydrothermal vent ecosystems in the
deep ocean; it's called "Vent Polymerase."
The
Taq
Polymerase is put with the DNA to be amplified, plus all four NTPs,
plus
two primers facing each other, about 200 - 6000 kb apart. (Why
do we need primers?) The primers are
selected based on the DNA region you want to amplify. The tube is
placed in a thermal cycler.
DNA
gets
synthesized from each primer, for about 2 minutes. Then the
temperature
is raised to 95 degrees C -- enough to denature (split apart) the DNA
base
pairs. But the Taq Polymerase remains intact, because it comes
from
an organism that evolved to grow at this temperature.
Now
the
temperature is decreased again, and primers again can hybridize to the
DNA--both the old AND the newly synthesized strands. Again, Taq
Polymerase
extends new DNA strands. Again, the temperature is raised.
After
repeated
cycles, the amount of DNA sequence between
the two primers increases exponentially.
First 2 strands, then 4, 8, 16, up to about a million. Thus, in a
couple of hours, you can get million-fold amplification of a DNA
sequence.
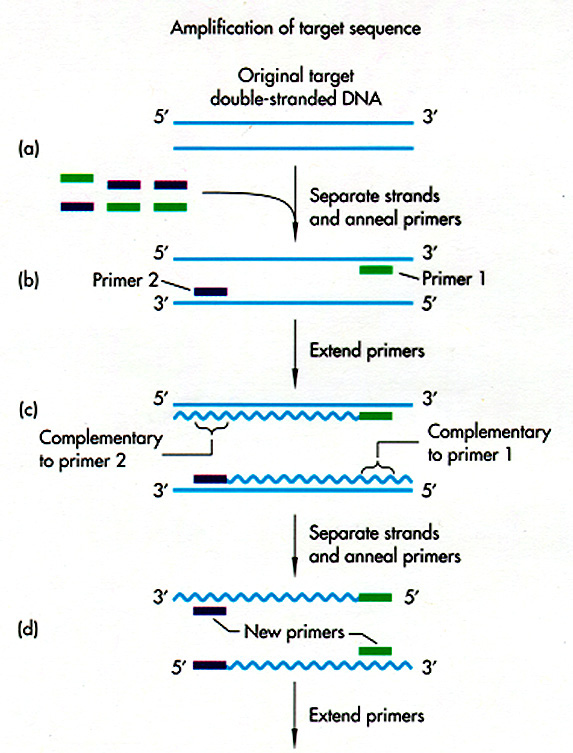
Griffiths
et al, W. H. Freeman & Co., current edition
Applications
of
PCR
PCR has replaced cloning
for
many purposes, particularly the sequencing of DNA. It is faster
and
requires no vectors, which can mutate as they reproduce. It
can be used forensically, to amplify tiny amounts of DNA from criminal
evidence; or clinically, to detect DNA sequences linked to inherited
disorders.
PCR
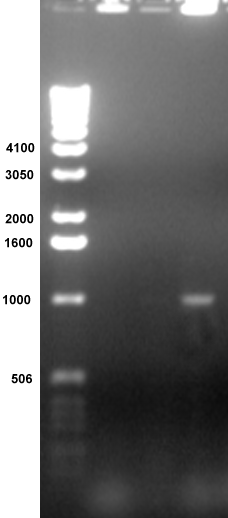
Experiment
in
Microbiology
Lab:
Students
characterized
the microbial species of various environments in Knox County.
Microbial
colonies
were placed directly into PCR reactions containing primers for
amplification
of ribosomal RNA genes (rDNA).
Lane
4 contains an
amplified band of the predicted size, 1000 bp. The top of each
well
contains genomic DNA. The smears at the bottom contain the PCR
primers.
The
DNA will be sequenced
and matched through GenBank to determine the microbial genus.
Thanks
to Dan Nickerson
'00 and Adam Marks '01.
|
 |
The main limitations of
PCR are:
-
Only relatively short
sequences can
be amplified reliably. Anything more than 10,000 base pairs is
unlikely
to be amplified.
-
You need to know the
right primer
sequences to use, at both ends of the sequence you want to
amplify.
If two related genes have the same end sequences, you might amplify the
wrong gene.
-
You only obtain a DNA
fragment.
To see this DNA at work inside a living organism, some type of cloning
has to be done.
For more information on PCR
see:
-
PCR
Technology - discussion of preparation of the sample, the master
mix,
and primers, and detection and analysis of the reaction products.
Gene
sequence analysis Modified from MIT
Hypertextbook.
The sequence of DNA base
pairs
can be analyzed by
-
Restriction
mapping. Construct a "road map" of
restriction
sites. A program to do this is WebCutter.
- DNA
Sequence
Analysis. Cut and clone various restriction
fragments, and determine the exact
sequence
of base pairs. All sequence information is deposited in GenBank.
If you just want the sequence of the peptide translated
from the RNA, you have to look for insulin mRNA
or
cDNA.
Once we have a piece of DNA
cloned, it is amplified (available in
many
copies) and we now have a living clone
which
provides, in theory, an indefinite source of the DNA sequence.
How
do we analyze the sequence?
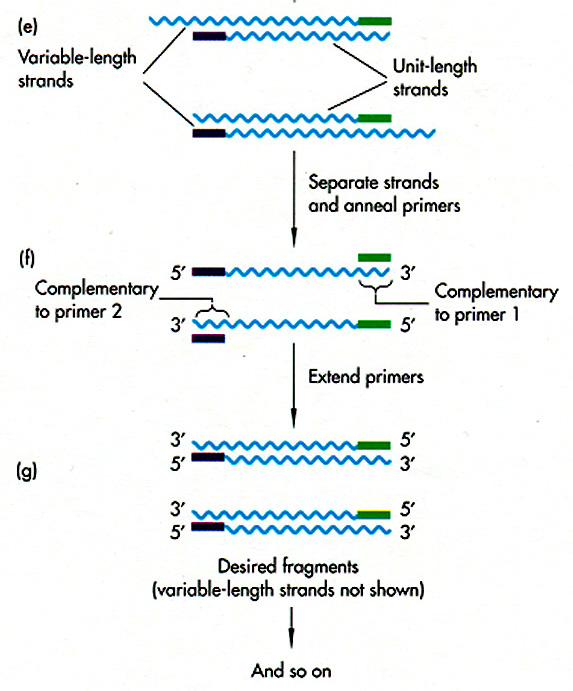
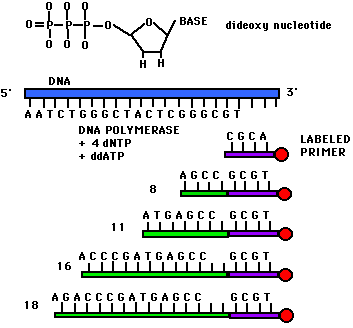
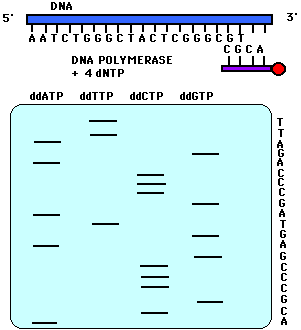
DNA Sequence Determination
Once you have identified a
particular
region of DNA of interest, you need to find out the precise sequence of
DNA nucleotides. This is done by di-deoxy
sequencing,
in which a DNA polymerase is put together with dNTPs in four different
reactions, each containing a small amount of one di-deoxy NTP (ATP,
TTP,
CTP, or GTP). The di-deoxy nucleotide lacks a 3'OH to continue
chain
extension, so the chain terminates. Each reaction produces a
population
of fragments terminated at A, T, C, or G.
The fragments are
either radiolabeled
or enzymatically labeled. They can be separated on a gel, or on a
fluorescence analyzer. All published DNA sequences in the world
are
deposited in GenBank.
images from Davidson
College
| Historically,
the products
of dideoxy sequencing reactions were subjected to electrophoresis in
four
different lanes of a gel. Each lane contained a reaction using a
different dideoxy terminator nucleotide. The data looked
like
the image at right. |
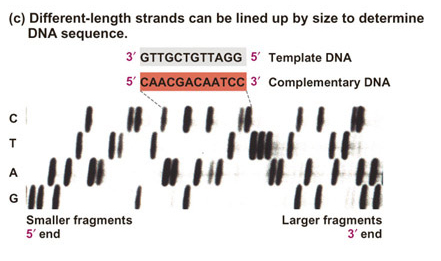
Image from your textbook, Freeman,
S. (2002) Biological Science. |
Today,
products of all four
sequencing reactions are loaded in a single gel lane or capillary tube
and subjected to electrophoresis. Molecular labels consist of
fluorescent
dyes instead of radioactive nucleotides. The gel looks something
like this:

Image from the University
of Maine DNA Sequencing Facility. |
Sequences of
DNA in the gel
lanes are read by a computerized fluorescence detection system that
measures
the intensity of light emission from each "band." The final
output is a "trace" or "electrophoretogram" that plots the intensity of
different color emissions vs. the length of the DNA being
sequenced.
By observing the progression of peaks of different colors, the DNA
sequence
is derived (A C
G T). The processed data look
like
this:
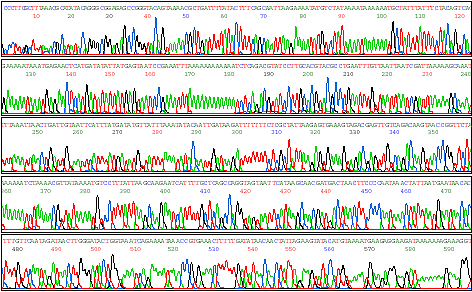
Image from http://www.qiagen.com/ |
Complementarity
and Hybridization
How do we find genes of
interest
in a genome? Or a particular mRNA in the total cellular
RNA?
Or a particular protein out of all cell protein?
In solution, hybrid
molecular
complexes (usually called hybrids) of the following types can exist
(other
combinations are possible):
-
DNA-DNA.
A single-stranded DNA molecule (ssDNA probe)
can form a double-stranded, base-paired hybrid with a ssDNA target if
the
probe sequence is the reverse complement of the target sequence.
A radiolabeled DNA probe can be applied to DNA from a gel transferred
to
a membrane, called a Southern Blot
(named for its inventor).
-
DNA-RNA.
A single-stranded DNA (ssDNA) probe molecule can form a
double-stranded,
base-paired hybrid with an RNA (RNA is usually a single-strand) target
if the probe sequence is the reverse complement of the target
sequence.
An RNA can be radiolabeled to probe a Southern Blot; or, a ssDNA probe
can be applied to membrane-bound RNA, called a Northern
Blot (name is a pun on Southern.)
-
Protein-Protein.
An antibody probe molecule (antibodies are proteins) can form a complex
with a target protein molecule if the antibody's antigen-binding site
can
bind to an epitope (small antigenic region) on the target protein. In
this
case, the hybrid is called an 'antigen-antibody complex' or 'complex'
for
short. A radiolabeled antibody can probe membrane-bound proteins,
called a Western Blot
(an even worse pun.)
There are two important
features
of hybridization:
-
Hybridization
reactions are specific - the probes will only bind to targets
with
complimentary sequence (or, in the case of antibodies, sites with the
correct
3-d shape).
-
Hybridization
reactions will occur in the presence of large quantities of molecules
similar
but
not identical to the target. That is, a probe can find one molecule of
target in a mixture of zillions of related but non-complementary
molecules.
These properties allow you
to use
hybridization to perform a molecular search for one DNA molecule, or
one
RNA molecule, or one protein molecule in a complex mixture containing
many
similar molecules.
Southern,
Northern, and Western Blots. Link to summary.
Blots
are named for
the target molecule.
Southern Blot--DNA cut with
restriction
enzymes - probed with radioactive DNA.
Northern Blot--RNA - probed
with
radioactive DNA or RNA.
Example--used to measure angiopoietin angiopoietin
expression from cDNA in transgenic mouse.
Western
Blot--Protein
- probed with radioactive or enzymatically-tagged antibodies.
These molecules must
then be immobilized
on a solid support, so that they will remain in position during probing
and washing. The probe is then added, the non-specifically bound probe
is removed, and the probe is detected. The place where the probe is
detected
corresponds to the location of the immobilized target molecule. This
process
is diagrammed below:
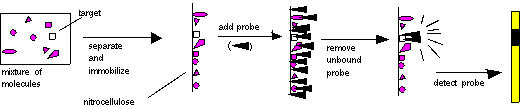
In the case of Southern,
Northern,
and Western blots, the initial separation of molecules is done on the
basis
of molecular weight, by gel electrophoresis.
-
Preparing
for Blots
-
Southern Blots.
DNA is first
cut with restriction enzymes and the resulting double-stranded DNA
fragments
have an extended rod conformation without pre-treatment.
-
Northern Blots.
Although RNA
is single-stranded, RNA molecules often have small regions that can
form
base-paired secondary structures. To prevent this, the RNA is
pre-treated
with formaldehyde.
-
Western Blots.
Proteins have
extensive 2' and 3' structures and are not always negatively charged.
Proteins
are treated with the detergent SDS (sodium dodecyl sulfate) which
removes
2' and 3' structure and coats the protein with negative charges.
Transfer
to Solid Support. After the DNA, RNA, or
protein
has been separated by molecular weight, it must be transferred to a
solid
support before hybridization. (Hybridization does not work well in a
gel.)
This transfer process is called blotting and is why these hybridization
techniques are called blots. Usually, the solid support is a sheet of
nitrocellulose
paper (sometimes called a filter because the sheets of nitrocellulose
were
originally used as filter paper), although other materials are
sometimes
used. DNA, RNA, and protein stick well to nitrocellulose in a
sequence-independent
manner.
After a series of treatment
steps,
the probe is added. The probe hybridized to the target
molecules
is visualized either by autoradiography or by enzyme reaction.
Summary.
The important properties of the three blotting procedures of DNA
analysis:
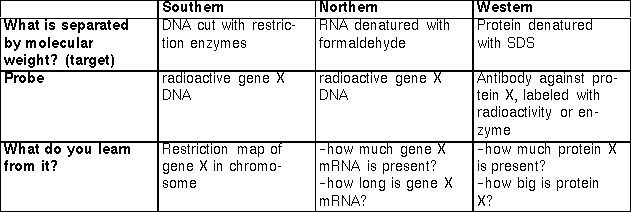
DNA
Microarrays
We
can now
put most of the protein-encoding genes onto a microarray chip, using
technology
based on the DNA silicon chip industry. The chip can be used to
hybridize
to cellular RNA, and measure the expression rates of a large number of
genes in a cell.
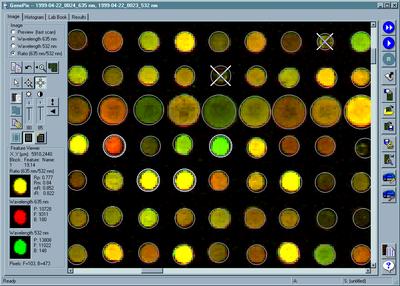
Axon Industries.
From "Everything's Great When
It
Sits on a Chip," The Scientist, Volume 13, #11, May 24, 1999
Transgenics
Cloning
in Animals.
Animals have two different
classes
of cells: germ line and somatic.
Only alterations in the
germ
cells can be transmitted to future generations. However, some
forms
of somatic cell gene therapy
can be useful in treating patients. For example, people with
cystic
fibrosis can receive the cloned CFTR gene in a nasal spray, which then
infects the lining of their lungs and improves lung function. The
infected (transformed) epithelial cells eventually are lost, however,
during
normal processes of tissue growth, so the treatment needs to be
repeated.
Germ-line
cloning.
There are two basic ways to clone a gene in the mammalian germ line.
-
Inject a DNA fragment
containing
your gene into the nucleus of a fertilized egg (germ-line
gene cloning) or of body tissues in a mature host (somatic
gene cloning). The DNA gets taken up at random somewhere
in
one of the chromosomes.
An example of germ-line
cloning is the injection of angiopoietin
cDNA into the fertilized mouse egg.
(An example of somatic
cloning is gene therapy for cystic fibrosis: inhale a vector containing
the CF gene, which gets incorporated into the DNA of cells lining the
lungs.
Somatic cloned genes are not inherited by offspring.)
- Put a transgene
containing positive
and negative selective markers into an embryonic
stem cell tissue culture. The ES
cells
take up the gene by homologous recombination, replacing the host
allele.
The ES cells now are injected into a blastula of an unrelated host, and
some of the next generation progeny arise from the ES cells. To
learn
more about this, read Capecchi's article
on
Targeted Transgenes, on reserve.
Once
you have
cloned a valuable gene into an animal, say a sheep that makes insulin
in
its milk, how can we produce lots of identical progeny quickly without
sexual reproduction? This is what the popular press calls
"cloning."
(More after spring break.)
But when you create a
clone, how
do you know it worked? You need to
know:
-
DNA:
Did the
transgene really incorporate into the genome? Where?
-
RNA:
Is the
RNA expressed? (Or prevented from expression, by a null allele?)
-
Protein:
Is
a desired protein expressed?
Cloning in
Plants.
Plants don't have separate
somatic
and germ cells, so creating transgenic plants is easier than creating
transgenic
animals. Techniques include the use of microprojectile bombardment (the
"gene gun") and the use of Agrobacterium tumefaciens.
Online
practice problems: Try problems 1-9 of the Recombinant
DNA Technology Problem Set from
the Biology Project at
the
Univerisity of Arizona.
|
