Unique as a pathogenic agent, the cause of Creutzfeldt-Jakob disease (CJD) is believed to be a protein. This "proteinaceous infectious agent" was termed a prion by S.B. Prusiner, the theoretical father of this recently discovered disease phenomenon (Prusiner, 1982) [see an article by the man himself]. The prion diseases include all of those in which there is a build-up of of the abnormal isoform of the prion protein, PrP-SC, which usually leads to spongiform encephalopathies. Human prion diseases include CJD, kuru, Gerstmann- Straussler-Scheinker disease (GSS), fatal familial insomnia (FFI) and the animal diseases believed to be caused by prions are scrapie in sheep, bovine spongiform encephalopathy (BSE or "mad cow disease)" and transmissible encephalopathy in mink. Scrapie was the first prion disease to be discovered, however this soon led to the recognition that kuru, an illness of progressive cerebral ataxia transmitted by ritualistic cannibalism within the Fore people of Papua New Guinea, was also a prion disease. Due to its resemblance of scrapie and kuru, CJD was soon also included in this unique catalog of disease agents. Although the human prion diseases are all very rare, there has been significant recent hype over BSE due to instances of disease transmission to humans from the consumption of infected cow carcases.
There are several different forms of CJD, which are summarized below:
The most common prion disease in humans is sporadic CJD, of which less than 1% of these cases have been found to be infectious (Scott et al., 1996). In fact, numerous attempts to prove that sporadic prion diseases are even caused by infection have failed; hence, sporadic CJD is likely caused by spontaneous conversion of PrP-C to PrP-SC within an individual. Consistent data throughout many countries supports the incidence rate of one case per million population per annum, providing further proof for the liklihood that sporadic CJD is caused by a spontaneous mutation. Nonetheless, prion diseases may be inherited as well - between 10 and 15% of prion disesases are passed on as an autosomal dominant genetic trait (Scott et al., 1996).
Although the liklihood of acquiring CJD is extremely rare, (Davanipour
et al., 1985) attempted to identify risk factors for the disease. They
concluded from case-control studies that head or neck trauma, surgery requiring
stitches, opthalmic exams using a tonometer, contact with wild animals,
and eating pork or rare meat could all increase one's chances of getting
CJD. Moreover, those afflicted individuals had more than average exposure
to wild animals such as deer and rabbits. Several cases of CJD have been
traced to tissue transplants or injection of foreign body material (check
out --advisory
report to Canadian blood donors). Corneal and dura matter transplants
as well as foreign pituitary growth hormone injection have all been cited
as origins of transmission (Steelman, 1944).
Of course there have also been cases of transmission of related neurological
degenerative prion diseases to humans from ingestion of infected meat.
In actuality, there is an extremely minimal threat of getting a prion disease
via this route. the
risk from eating beef less than driving car or having sex". CJD poses a
serious epidemiological concern to hospitals, according to Steelman
(1994) who highlights the need to protect hospital workers and other
patients from the causative disease agent. It is resistive of routine sterilization,
disinfection, and cleaning techniques and therefore requires increased
precaution.
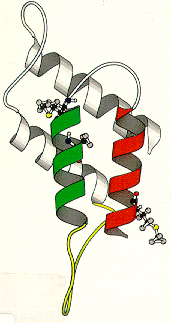
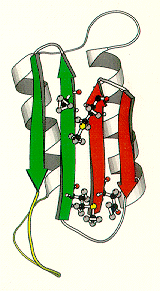
"small proteinaceous infectious particles that resist inactivation by procedures modifying nucleic acids and contain an abnormal isoform of a cellular protein which is a major and necessary component."these diagrams are from an article by Huang, Prusiner, and Cohen, (1996) On the left is the normal prion protein, PrP-C. It consists of approximately 45% alpha-helix and lacks almost entirely any beta sheet. On the right is the abnormal conformer, PrP-SC. It consists of approximately 30% alpha-helix and 45% beta sheet (Pan et al., 1993). PrP-C appears to be post-translationally modified to PrP-SC only in conformation by the acquisition of increased beta sheet conformation and loss of some alpha-helix.
A wide variety of studies have been dedicated to researching the nature of the infectious prion particle. They have included methods of molecular engineering of transgenic animals, neuropathological exams, as well as biochemistry, immunology and cell biology investigations. The bulk of research has led to the theory that either PrP is entirely responsible for disease pathogenesis or that as a result of the pathogenic process, PrP accumulates in infected material. This is an ongoing and current field of study; new variants of CJD are still being discovered.
The phenomenon of prion replication has raised numerous previously unencountered questions. The Central Dogma of biological theory requires that nucleic acid act as a template for replication. However prions violate this theory if they are made up of negligable amounts of nucleic acid. Faithful believers in the power of prions have attempted to explain possible methods of prion replication -- the prion dimer hypothesis claims that an initial PrP-SC molecule combines with one wild-type PrP-C to form a heterodimer (Prusiner, 1982). This is then converted into a homodimer (of PrP-SC/PrP-SC) that could then dissociate and lead to further conversion to the pathogenic agent. From the initial presence of PrP-SC, there would be an exponential increase in the abnormal conformer. The gene to produce PrP-SC could therefore be genetically passed on or an individual could acquire some of the abnormal protein from an infected individual (such as the manner in which BSE was passed onto humans). Otherwise, the normal PrP-C gene could mutate to produce PrP-SC or the normal protein could spontaneously change to form the abnormal protein. Of course the liklihood of either event occurring is very low, but this is believed to be the cause of the rare sporadic CJD.
There is also counter-evidence to the theory that CJD is caused by prions. Sklaviadis and collegues (1989) found that the CJD infectious agent is larger in size than what would be expected for a monomeric protein. From density gradient resolution, it also was shown to have a greater density than other proteins resolved and detected in the same gradient. This study concludes that the CJD pathogenic agent is more likely to be similiar to conventional animal viruses than solely a protein complex, due to the detection of a structure consisting of protein as well as significant nucleic acid.
So just how does the abnormal PrP isoform cause the neurological degeration observed in individuals afflicted with CJD? This aspect of research has not been fully elucidated, although several interesting studies have investigated prion pathogenesis. It has been shown that in vivo, CNS neurons express the highest amount of PrP mRNA (Prusiner, 1982). In addition, when transgenic mice were constructed to overexpress PrP-C and then samples of their brains were removed and transmitted to hamsters, the hamsters developed scrapie-like brain infection (Prusiner, 1996). The titer of scrapie infectivity was found to be proportional to PrP-SC concentration. This evidence supports the hypothesis that PrP-C may be spontaneously converted to the pathogenic PrP-SC, which causes symptomatic neurological disorder. At this point, the increasing amount of PrP-SC complexes aggregate together and are resistant to normal degredation by proteases. PrP-C has been found to be completely digested by limited esposure to proteinase K, whereas PrP-SC is only partially digested and even still retains its infectious nature (Westaway et al., 1994). The resistance of PrP-SC to proteolysis may also enhance the rod formation and subsequent neurodegeneration. Apparently the presence of the beta sheet and the lower concentration of the alpha-helix conformation in PrP-SC compared to PrP-C increases the propensity of the protein to aggregate into rods. Amyloid plaques which stain with alpha-PrP antibodies serve as a diagnostic tool for CJD identification (Prusiner, 1982). Not all patients with CJD have these plaques; often familial CJD cases may just present with widespread spongiform degeneration.

Richardson, E.P.J. (1977) Myoclonic dementia: introduction IN Neurologic Classics in Modern Translation (ed. D.A. Rottenberg and F.H. Hochberg) New York: Hafner Press.
Davanipour, Z. et al. (1985) Creutzfeldt-Jakob disease: possible transmission by consumption of wild animal brains. Neurology 35:1483-1486.
DeArmond, S.J. and Prusiner, S.B. (1995) Am. J. Path. 146(4):785-811
Gajdusek, D.C. (1991) The transmissible amyloidoses: genetic control of spontaneous generation of infectious amyloid proteins by nucleation of configurational change in host precursors: kuru-CJD-scrapie-BSE. Eur. J. Epidemiol. 7(5):567-577.
Huang, Z., Prusiner, S.B. and Cohen, F.E. (1996) Structures of prion proteins and conformational models for prion diseases IN Prions Prions Prions (ed. S.B. Prusiner) Berlin: Springer-Verlag.
Oken, R.J. and McGeer, P.L. (1995) Human prion diseases: possible new directions in prophylaxis and therapy. Medical Hypotheses 44:167-168.
Pan, K.M. et al. (1993) Conversion of alpha-helices into beta sheets features in the formation of scrapie prion proteins. Proc. Natl. Acad. Sci. USA 90:10962-10966.
Prusiner, S.B. (1991) Molecular biology of prion diseases. Science 252:1515-1552.
Prusiner, S.B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136-144.
Prusiner, S.B. (1996) Human prion disease and neurodegeneration IN Prions Prions Prions (ed. S.B. Prusiner) Berlin: Springer-Verlag.
Scott, M.R.D. et al. (1996) Transgenetics and gene targeting in studies of prion diseases IN Prions Prions Prions (ed. S.B. Prusiner) Berlin: Springer-Verlag.
Sklaviadis, T.K., Manuelidis, L. and Manuelidis, E.E. (1989) Physical properties of the Creutzfeldt-Jakob disease agent. J. Virol. 63(3):1212-1222.
Steelman, V.M. (1944) Creutzfeld-Jakob disease: Reccomendations for infection control. Am. J. Infect. Control 22(5):312-318.
Westaway et al., (1994) Degeration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 76:117-129.