
Hamartomaceous lesions, or hamartomas, are tumour-like growths which
are referred to as tubers. The most common tuber forms of the internal
organs are cerebral hamartias and subependymal giant cell astrocytomas
of the brain, rhabdomyomas of the heart, and angiomyolipomas of the kidneys
(Kwiatkowski and Short, 1994 cited in Henske et. al., 1996). Lesions
of the brain occur at the gray-white matter interface and displace the
normal cortical tissue. 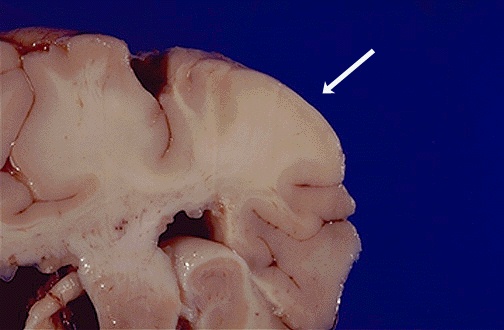 The presence of brain lesions is correlated with the occurence of mental
retardation and seizures, although only 60% of TSC patients have seizures
and only 40% are mentally retarded. Brain involvement has also been linked
to autism and behavioral disorders. Over 50% of children with TSC are autistic
and many also experience attention deficit disorder and hyperactivity.
Subependymal giant cell astrocytomas sometimes obstruct the flow of cerebrospinal
fluid and have to be removed surgically. As a TSC patient ages, the subependymal
lesions often calcify allowing easier detection by MRI or CT scans (Tuberous
Sclerosis).
The presence of brain lesions is correlated with the occurence of mental
retardation and seizures, although only 60% of TSC patients have seizures
and only 40% are mentally retarded. Brain involvement has also been linked
to autism and behavioral disorders. Over 50% of children with TSC are autistic
and many also experience attention deficit disorder and hyperactivity.
Subependymal giant cell astrocytomas sometimes obstruct the flow of cerebrospinal
fluid and have to be removed surgically. As a TSC patient ages, the subependymal
lesions often calcify allowing easier detection by MRI or CT scans (Tuberous
Sclerosis).
Children with TSC are sometimes affected by renal cysts, but adults
more often experience renal angiomyolipomas. Renal cysts are potentially
dangerous because they may lead to end-stage renal disease and hypertension.
Angiomyolipomas are generally not life-threatening, but in a small number
of cases renal lesions associated with TSC have developed into carcinoma
(Tuberous Sclerosis).
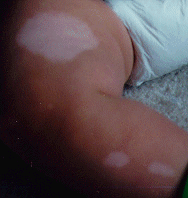
A variety of dermatological disorders have been correlated to TSC. Some of these include hypomelanotic macules, confetti skin lesions, facial angiofibromas, ungual fibromas, Shagreen's patches, and forehead plaque. Hypomelanotic macules have been found, usually with a Wood's light, in patients of all ages and even in fetuses. These macules can be polygonal or Ash-leaf shaped or can occur in groups of small white spots.
Facial angiofibromas, or Adenoma Sebaceum, are one of the most distinctive
indicators of TSC. 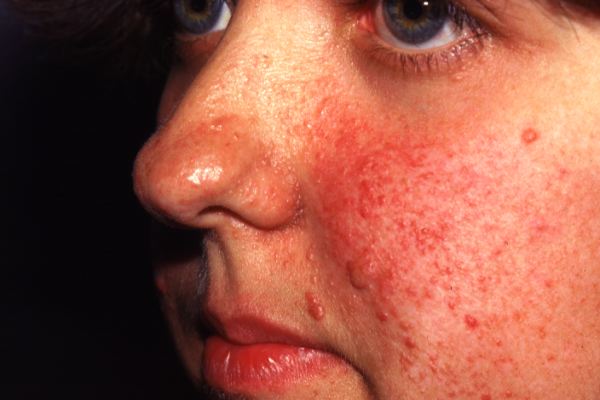 These reddish papillary lesions are found around the nose, cheeks, and
chin and are present in 70% of people affected by TSC. These lesions usually
develop between five years of age and puberty. Ungual fibromas are also
found on the face, near the forehead, eyelids, cheeks, or scalp and can
be present from birth (Australasian
TS Society).
These reddish papillary lesions are found around the nose, cheeks, and
chin and are present in 70% of people affected by TSC. These lesions usually
develop between five years of age and puberty. Ungual fibromas are also
found on the face, near the forehead, eyelids, cheeks, or scalp and can
be present from birth (Australasian
TS Society).
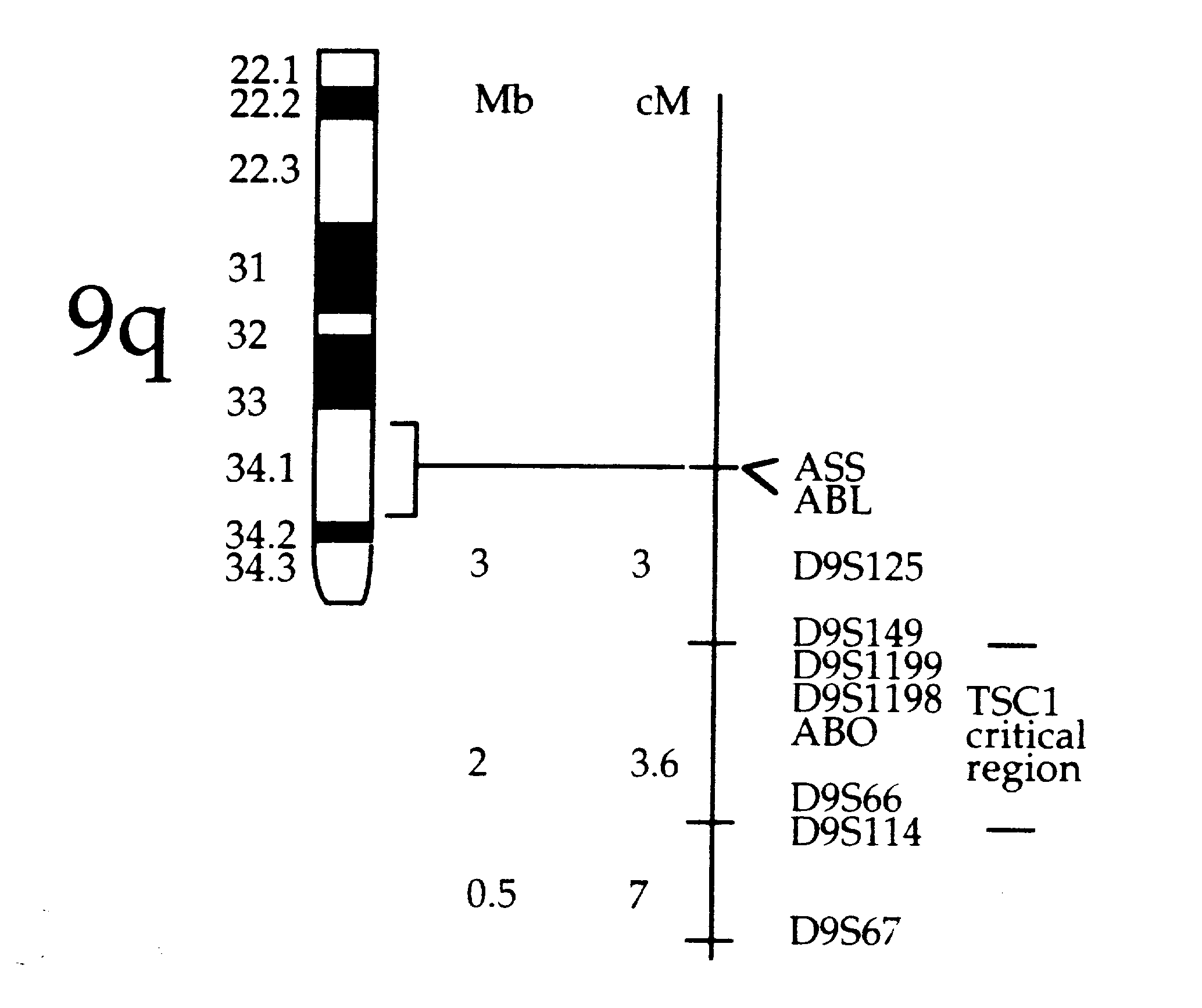
Tuberous Sclerosis Complex is an autosomal dominant disorder caused by a mutation in one of two known genes. The TSC1 gene is located on chromosome 9q34 and the TSC2 gene on chromosome 16p13. The precise location of the TSC2 gene was found in 1993 and it is now known to be responsible for the production of the protein tuberin which suppresses the growth of tumors. Mutations in this gene prevent the production of tuberin and allow certain tissues to grow unchecked. Researchers are still trying to determine the location of the TSC1 gene on chromosome 9 (Tuberous Sclerosis Newsletter).
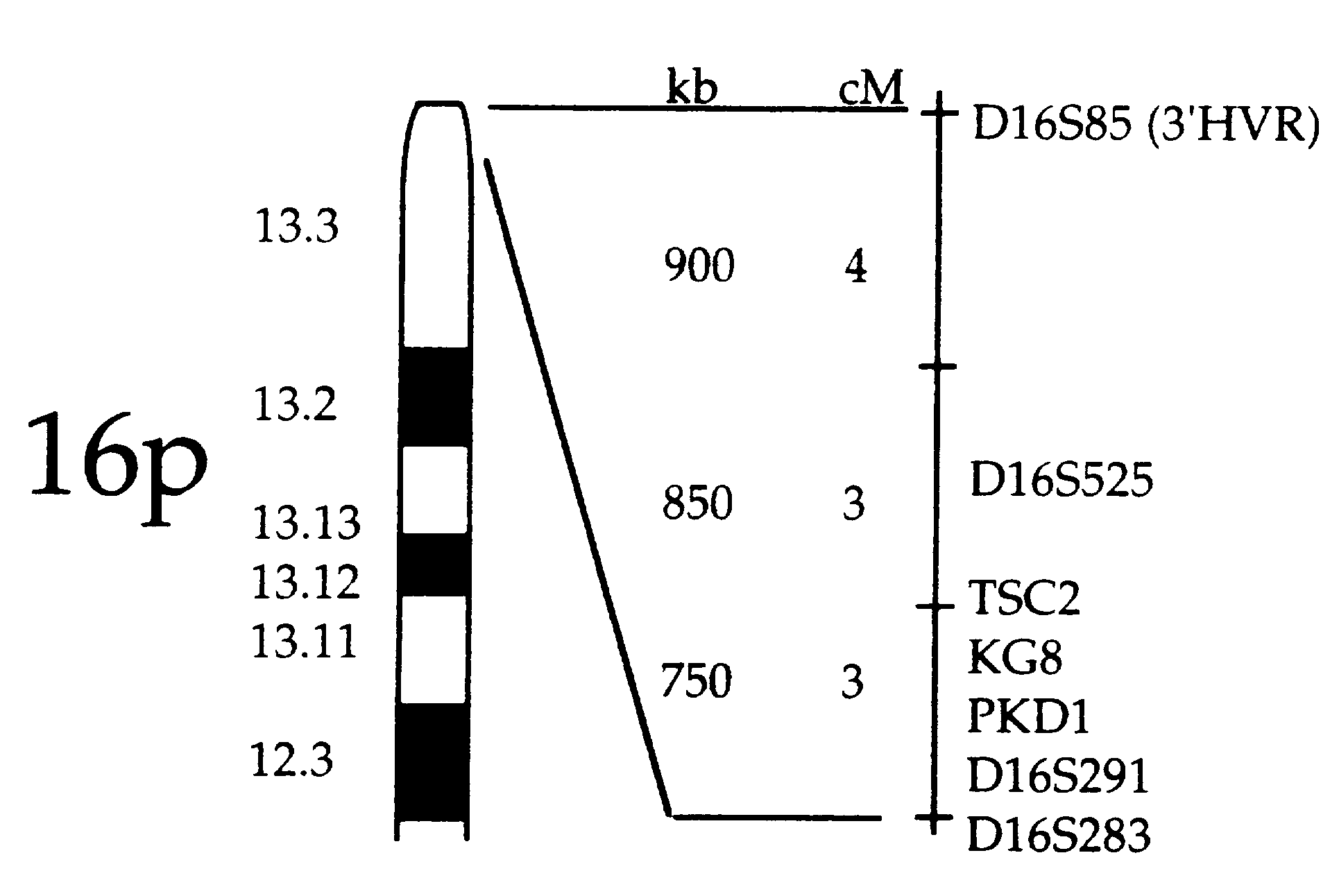
Past linkage studies involving the TSC genes have indicated that there is a nearly equal division of families having mutations in the TSC1 and TSC2 genes. Analysis of linkage to certain markers and recombination data were used to locate TSC2 and are currently being used to study TSC1. These efforts are complicated by small sample sizes, conflicting information within the same families, and the lack of knowledge about how the TSC genes work (Tuberous Sclerosis).
Several cases of identical twins exhibiting differing degrees of TSC have been recorded. There are many hypotheses about the cause of this discordancy of expression, however, studies have not conclusively proven any of them. One of the most widely accepted hypotheses for this phenomenon is imprinting of the TSC genes. Theoretically, if one embryo received more imprinted cells than the other during division, the two children could have differing degrees of TSC. Although there is no real evidence to support this theory yet, imprinting seems to be a promising new area of TSC research (Tuberous Sclerosis).
Cloning of the TSC2 gene has shown that it is partially homologous to the RAP1 GTP-ase-activating protein. Other studies are currently being conducted to investigate the loss of heterozygosity in the hamartomas of TSC patients. This is done by analyzing methylation patterns of a segment of the X-chromosome in order to determine whether or not it is activated. Once the methylation pattern is determined, it can be extrapolated to fit the TSC genes as well. One such study, conducted by Green et. al., 1996 on female TSC patients, showed that in hamartomaceous lesions the inactivation pattern is scewed so that one X-chromosome is fully methylated and the other is unmethylated, instead of showing the random pattern of inactivation seen in normal tissue. This validates the hypothesis that the TSC genes are involved in tumor suppression, because inactivation of both copies of a gene causes a loss of growth control in a cell which allows it to proliferate and form a tuber.
Another study by Henske et. al., 1996, also investigated loss of heterozygosity in lesions, focusing on the difference between the TSC1 and TSC2 genes and the different organs where the lesions are found. Testing of lesions from 47 patients revealed that loss of heterozygosity is more frequently seen in relation to TSC2 than TSC1. The researchers also found that loss of heterozygosity was seen in 56% of renal and cardiac lesions but in only 4% of brain lesions. The study concluded that brain lesions must form by a different mechanism that other hamartomas.
The search for specific mutations which lead to TSC is a major topic of current research. A paper by Vrtel et. al., 1996, identified a point mutation which caused TSC in a father and son. A genomic phage library of the TSC2 gene was prepared and analyzed by polymerase chain reaction (PCR) and single strand confirmation polymorphism (SSCP) analysis. The mutation was detected in the first exon of the 5' end of the gene. Several large deletions were detected in other patients as well. The relatively mild cases of this father and son and the lack of tumors in the family suggested that perhaps this mutation only caused a partial loss of tuberin producing ability.
Geneticists are optimistic about the future of TSC research. The recent mapping of the TSC2 gene has opened an entirely new area of research which focuses on determining which mutations are responsible for the disease. Researchers are quickly closing in on the location of TSC1 as well, and this new information will hopefully lead to even better diagnostic tests and better treatments for those who suffer from this disease.
Henske, E.P., B.W. Scheithauer, M.P. Short, R. Wollmann, J. Nahmias, N. Hornigold, M. van Slegtenhorst, C.T. Welsh, and D.J. Kwiatkowski. 1996. Allelic loss is frequent in Tuberous Sclerosis kidney lesions but rare in brain lesions. Am. Journal of Human Genetics 59: 400-406.
Vrtel, R., S. Verhoef, K. Bouman, M. Maheshwar, M. Nellist, A. Essen, P. Bakker, C. Hermans, M. Bink-Boelkens, R. Elburg, M. Hoff, D. Lindhout, J. Sampson, D. Halley, and A. Ouweland. 1996. Indentification of a nonsense mutation at the 5' end of the TSC2 gene in a family with a presumptive diagnosis of tuberous sclerosis complex. J. Med. Genet. 33: 47-51.