Antiviral agents are less well developed than antibacterial agents because most viruses are obligate intracellular parasites that require the host cell mechanisms for replication. Because of this, it is more difficult to create drugs which target the viral replication without damaging the host. The target of antiviral drugs depends on the type of virus involved; each antiviral therapy is different for each virus because of the different mechanisms of infection a virus induces. Viral DNA polymerase is a prime target of antiviral therapies because it is essential for viral replication. An ideal antiviral therapy works exclusively by inhibiting a viral-specific function and thereby causing minimal disturbance to the host cell metabolism (Miller, AHMF).
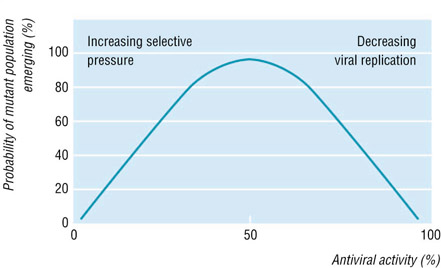
Drug resistance is defined as a reduced susceptibility to a drug in a laboratory culture system and is expressed as an altered IC50 or IC90 (drug concentration required to inhibit viral growth by 50% or 90%, respectively). This "phenotype" is determined by specific mutations in the viral genome, which leads to alterations in the viral drug activator (i.e. herpes simplex thymidine kinase). Sometimes, multiple mutations are required for the development of high level resistance, and insufficient suppression of viral replication by antiviral therapies will predispose this. The drug resistant virus is considered to be the fittest in the presence of the drug (see figure below). Antiviral therapies may fail due to poor drug compliance, pharmacological factors, and drug resistance (Pillay and Zambon).
 |
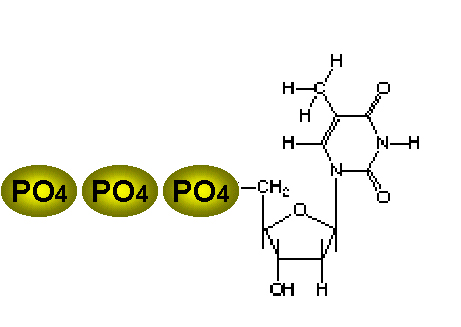
Thymidine |
The best antiviral therapies are selective because the virus may use its own enzyme to activate the drug, and/or the viral polymerase may be much more sensitive to the drug than the corresponding host enzymes. Thymidine kinase is an enzyme encoded by some viruses (i.e. herpes simplex virus) and used in the synthesis of their DNA. TK has the ability to activate certain drugs so that there is selectivity inducing the virus to activate the drug to a toxic form, initiating successful suppression of the infection, while the uninfected cell does not have this ability because it lacks TK. TK allows the virus to grow in cells lacking a high concentration of phosphorylated nucleic acid precursors, such as cells nerve cells that do not replicate their genome. Thymidine is the best nucleoside substrate for this enzyme (seen at left). |
Antiviral therapies such as acyclovir (ACV), valaciclovir (VAL), and famciclovir (FAM) are widely used to treat herpes simplex infections (HSV-1 and HSV-2, ACV, VAL, and FAM). In general, drug resistance is limited to immunocompromised patients; the estimated prevalence of herpes simplex resistant to ACV is 5-10% in patients with AIDS and 18% in recipients of bone marrow transplants (Pillay, 1998). HSV resistance to antiviral therapies occurs as a result of mutations in the virus-specific genes responsible for the action of ACV, namely, thymidine kinase and viral DNA polymerase. This occurs via three mechanisms: 1) mutant viruses deficient in TK; 2) mutants with altered substrate specificity of thymidine kinase; or 3) mutants with altered viral DNA polymerase enzymes. The most frequently observed resistant strains are unable to express TK and are avirulent in animal models of disease. Resistance may be acquired less frequently by selecting TK- or DNA polymerase-encoding variants which no longer recognize ACV or ACV triphosphate, respectively, as substrates but which retain normal functions. Clinical resistance to ACV is defined as the progression of mucocutaneous HSV lesions despite ACV therapy (Pielop et al., 2000). Nonsense, frame shift, and missense mutations can give rise to viral TK deficiency (Pillay, 1998). When an HSV lesion does not disappear after low-dose treatment, the first response should be to increase the dosage, which usually overcomes partial resistance (see figure below).

Management of ACV-Resistant HSV |
One study in England concluded that 0.1-0.6% of HSV isolates from immunocompetent hosts were resistant to ACV, while 6% of immunocompromised isolates were resistant, mostly in patients who had previously received antiviral therapy treatment. Another study conducted in the United States determined that 4.7% of immunocompromised and 0% of immunocompetent hosts with HSV expressed antiviral resistance to ACV in vitro (Pielop et al., 2000). An important fact is that many TK-deficient HSV mutants have been isolated and do not consistently result in antiviral resistance.
For patients that express ACV-resistant HSV
strains, other antiviral therapies are available. Some of these antivirals
are intravenous foscarnet, which does not require HSV-encoded TK to be
functional. This is the only FDA approved treatment for immunocompromised
patients with HSV. Other treatments not yet FDA approved are vidarabine,
which also is not dependent on TK activity and less effective than foscarnet,
and cidofovir, which is phosphorylated by cellular enzymes, thus inhibiting
viral DNA polymerase. Cidofovir is also ineffective against rare
strains with viral DNA polymerase mutants (Pielop
et
al., 2000).
Home
Previous
Next