The Human Prion Protein in
Complex with Nanobody 484
Carter Brzezinski '20 and Mackenzie Bruzzio '20
Contents:
I. Introduction
II. General Structure
III. N-terminal Domain
IV. Nanobody 484
VI. References
I. Introduction
Prion proteins are fatal neurodegenerative
transmissible agents which cause incurable illnesses known as
transmissible spongiform encephalopathies (TSEs). These diseases
arise when the cellular form of the human prion protein (HuPrPC)
undergoes several structural conversions and misfolds into its
oligomeric form know as the scrapie prion or HuPrPSc. The
misfolded human prion is most notbably the cause of
CreutzfeldtJakob disease (CJD) in humans. This disease is
associated with personality changes, anxiety, depression, and memory
loss, and is usually fatal. In its cellular form the HuPrP acts as a
synapse buffer and mediates copper ion concentration between
neurons. Although this interaction is important, it is not
completely understood.
The HuPrPSc is insoluble in most organic solvents, partially
resistant to Proteinase K, and prone to aggregation. This causes
problems when trying to isolate the prion for x-ray crystallography.
However, when in complex with nanobody 484, a nanobody with high
affinity for the human prion, an ultra-clear crystal is able to
form. Further understanding of the structure of the prion will lead
to both understanding of the prions normal cellular function as well
as the mechanism to which the cellular form folds to the scrapie.
Once this mechanism is understood, a functional treatment for the
fatal diseases associated with this protein can be formulated and
the role of its cellular type can be solved.
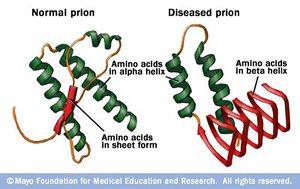
II. General Structure
HuPrPC has two
. These are known as the Globular
domain and the N-terminal tail
. The Globular domain consists of
and three short anti-parallel
The addition of Nb 484 to HuPrP causes enrichment of the three
β-strands, but more notably the expression of the
This "hairpin turn" includes a palindromic GGLGG
, and is is directly exposed to solvent. Specifically,
are exposed and create a dry hydrophobic pocket that allow
for intermolecular β-strand stacking in either parallel or
antiparallel configuration. Thus, the expression of the β0-β1
hairpin caused by the binding of Nb 484 appears to instigate the
interaction with intermolecular β-strands to form steric zippers
and contributes to the inhibition of HuPrP misfolding.
III. N-terminal Domain
Two papers by Hornshaw et al. suggested that PrPC binds Cu2+ in
its so called octarepeat domain. This domain is composed of
multiple repeats of the eight-residue sequence PHGGGWGQ. The
HuPrP sequence carries four copies of this segment. This region
is unstable when treated with Proteinase K, and therefore is not
shown in the core model. In fact, the
(displayed in this pdb as a short sequence) is
actually a long chain with multiple binding domains as shown by
the image below.

The octarepeat domain in HuPrP spans position 70 to position
94. As found by Wopfner et. al., across different species the
octarepeat domain is highly conserved within the PrP sequence.
This suggests that it plays a role in a prions cellular
function. As shown in Figure 2, one octarepeat domain is able to
coordinate a single Cu2+ ion. This equatorial coordination is
mainly done by the histidine imadazole, deprotonated amides from
the glycines, and the amide carbonyl of the second glycine.
There are also 3 water mediated interactions.
 Millhauser reports that this sequence is extremely selective
towards Copper over other divalent ions due to Coppers unique
ability for acid-base interactions with the amide carbonyl of
glycine. These findings further suggest that the human prion
protein has something to do with Copper concentration mediation in
vivo. Interestingly, when the scrapie version of this protein is
cut with Proteinase K, therefore losing this octarepeat domain, it
does not lose its infectivity. From this we can conclude that the
ability of HuPrP to coordinate Copper does not play a role in the
development of TSEs.
Millhauser reports that this sequence is extremely selective
towards Copper over other divalent ions due to Coppers unique
ability for acid-base interactions with the amide carbonyl of
glycine. These findings further suggest that the human prion
protein has something to do with Copper concentration mediation in
vivo. Interestingly, when the scrapie version of this protein is
cut with Proteinase K, therefore losing this octarepeat domain, it
does not lose its infectivity. From this we can conclude that the
ability of HuPrP to coordinate Copper does not play a role in the
development of TSEs.
IV. Nanobody 484
is a nanobody generated by Abskharon et al. with a high
affinity to bind to both human and mouse prion proteins. Notably,
this nanobody is highly effective in inhibiting HuPrP propagation
into its misfolded form. When treated with Nb 484, MoPrP amyloid
seeding assays show that lag phase of fibrillization was extended
by 40 hours. This indicates that the interaction of Nb 484 with
the full-length MoPrP inhibits the formation of PrPSc-like
aggregates. While its not known exactly why it inhibits the
conversion, its likely due to steric hindrance and rigidity from
bonding at the
The nanobody forms 3
to the α2-β2 loop of the prion which gives the loop
significant rigidity. There are also 5 significant
between Nb484 and HuPrP that add to this structure.
Interestingly, organisms that exhibit more rigid prion α2-β2 loops
show a lower chance of prion-misfolding related disease. Futher
structural stabilization of this motif may represent an effective
way for Nb 484 to fully inhibit prion misfolding.
V. References
Franziska Wopfner, Weidenhofer, G., Schneider,
R., von Brunn, A., Gilch, S., Schwarz, T. F., Werner, T., and
Schatzl, H. M. 1999. Analysis of 27 Mammalian and 9 Avian Prps
Reveals High Conservation of Flexible Regions of the Prion
Protein. J. Mol. Biol. 289:1163-1178.
Glenn L. Millhauser. 2004. Copper Binding in the
Prion Protein. Accounts of Chemical Research. 37:7985.
Martin P. Hornshaw, McDermott, J. R., Candy, J.
M., and Lakey, J. H.. 1995. Copper binding to the N-terminal
tandem repeat region of mammalian and avian prion protein:
structural studies using synthetic peptides. Biochem.
214:993-999.
Romany N. N. Abskharon, Giachin, G., Wohlkonig, A.,
Soror, S. H., Pardon, E., Legname, G., and Steyaert, J.. 2014. Probing
the N-Terminal β-Sheet Conversion in the Crystal Structure of the
Human Prion Protein bound to a Nanobody. J.A.C.S. 136:937-944
Back to Top