The
Sickle Cell Anemic Mouse
 Currently
the new development towards a cure or treatment of sickle cell anemia has
lead to the development of sickle cell anemic mice. From the late eighties
two labs, Ryan
et al. and Paszty
et al., have been working on projects to breed and develop mice which
have the same symptoms as humans with sickle cell anemia. These mice are
the break through for sickle cell anemia. With the mice more testing can
be done to examine the different treatment methods of sickle cell anemia
as well as the development of the disease from onset.
Currently
the new development towards a cure or treatment of sickle cell anemia has
lead to the development of sickle cell anemic mice. From the late eighties
two labs, Ryan
et al. and Paszty
et al., have been working on projects to breed and develop mice which
have the same symptoms as humans with sickle cell anemia. These mice are
the break through for sickle cell anemia. With the mice more testing can
be done to examine the different treatment methods of sickle cell anemia
as well as the development of the disease from onset.
Ryan et al. has worked on the construction of a mouse
model for sickle cell anemic traits that could be bred and used for research
purposes. Results from other researchers had shown that mice could be developed
which could make high amounts of human alpha and beta-globin using the
human hemoglobin genes inserted after the DNase
I super-hypersensitive sites (also called LCR
or locus control regions) injected into fertilized mouse eggs. Ryan
et al. used the same strategy to create mice with human alpha -globin genes
and human betaS or HbS globin. The mice that developed after
this injection were examined for the inserted DNA in the correct orientation
for transcription. Three progeny were found with head to tail tandem arrays
of the transgenes, which produced HbS protein. The mouse that had the highest
HbS expression was the initiator of a line of transgenic mice with HbS
expression.
Testing the HbS mice by deoxygenating the blood with
nitrogen gas showed no real effect of the human HbS gene in the mice. This
result would be expected because the mouse beta -globins are still being
produced and interacting with the HbS proteins to buffer against polymerization,
akin to the sickle cell carrier who shows no sickle cell symptoms. (Ryan,
1990)
To overcome the effect of the mouse beta-globin interactions
with the HbS the mice were bred with a beta-globin thalassemic (beta-thal)
mouse line to reduce the mouse beta-globin (mbeta-globin) levels. The HbS/beta
-thal mice produced were heterozygous for the transgenes and the beta-thal
mutation. mRNA levels were examined at the key developmental stages to
assure that the HbS was expressed at the correct stage of development.
Developmental progress of the disease is especially important in the mice
because treatments for sickle cell anemia will depend on those stages in
humans. The closer one mimics the condition in the experimental stage
the closer one can estimate the validity of a treatment or cure.
mRNA was also quantitated to determine the amount of HbS
being expressed verses the mbeta-globin. In both the HbS mice and the HbS/beta
-thal mice there was 3 to 5 times as much mRNA for the HbS than the mbeta
-globin mRNA indicating a higher human protein content than mouse protein.
For the human and mouse alpha-globin the mRNA amounts were similar.
Unlike the HbS mice erythrocytes that showed almost no
sickling traits approximately 90% of the HbS/beta -thal erythrocytes showed
sickling when treated with nitrogen gas to deoxygenate. HbS/beta-thal mice
also showed characteristic spleenic enlargements of 2 to 4 times wild type
size but are healthy otherwise. (Ryan
et al., 1990)
Ryan et al. and Greaves et al. both produced transgenic
mice with 50% HbS which mimicked the human sickle cell trait, but not the
debilitating disease itself. The sickle cell trait resemblance of the transgenic
murine erythrosiod cells was due to the expression of mbeta-globin and
malpha-globin in the mouse cells. Ciavatta et al. and Yang et al. both
deleted the mouse beta -globin genes and Paszty et al. deleted the malpha-globin
genes. These mice lines showed anemic conditions similar to human beta
and alpha thalassemia conditions and could be bred with transgenic mice
with only adult human hemoglobin production. (Ryan
et al., 1997)
Viability of these transgenic mice was a concern because
the mice would only have only human hemoglobin and it was known that human
fetal globin is a major factor in the survival of sickle cell disease patients
in utero. Human fetal hemoglobin (HbF) accounts for 70 to 90% of the hemoglobin
at birth. Since HbF has been proven to help control sickling of cells in
humans with sickle cell disease the worry was that the mice wouldnt have
that protection in utero and would die before birth. A new construct for
HbS globin was made with the HbF gene ahead of the HbS gene to assure the
survival of the mice in utero by assuring the switch from HbS to HbS after
birth (HbF/HbS). (Ryan
et al., 1997)
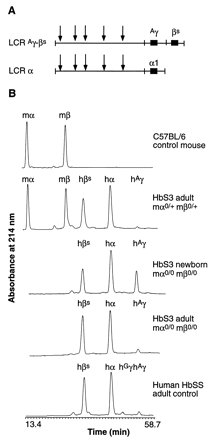 Mice
were bred to be heterozygous for m beta -globin, m alpha-globin and HbS.
These mice were crossbred to produce the lineage of mice homozygous for
the alleles without m beta -globin and m alpha -globin. These double knockout
HbF/HbS mice are similar to humans in that they have high HbF globin expression
at birth (approx. 30 to 50%) and "synthesize no murine hemoglobin" (Ryan
et al., 1997). These mice also persist to have a high but declining
HbF expression for a full month before the full hemoglobin switch to HbS
is completed. The figure to the left shows this switch clearly in the B
portion where a control mouse, HbS3 adult (heterozygous for the beta and
alpha mouse globin knowouts showing some mouse globin expression), the
HbS3 newborn with expression of only human hemoglobin and a similar adult
with only human adult hemoglobin beta and alpha being expressed.
Mice
were bred to be heterozygous for m beta -globin, m alpha-globin and HbS.
These mice were crossbred to produce the lineage of mice homozygous for
the alleles without m beta -globin and m alpha -globin. These double knockout
HbF/HbS mice are similar to humans in that they have high HbF globin expression
at birth (approx. 30 to 50%) and "synthesize no murine hemoglobin" (Ryan
et al., 1997). These mice also persist to have a high but declining
HbF expression for a full month before the full hemoglobin switch to HbS
is completed. The figure to the left shows this switch clearly in the B
portion where a control mouse, HbS3 adult (heterozygous for the beta and
alpha mouse globin knowouts showing some mouse globin expression), the
HbS3 newborn with expression of only human hemoglobin and a similar adult
with only human adult hemoglobin beta and alpha being expressed.
90% of the mice survived for 2 to 9 months, were fertile
and had many of the same symptoms of sickle cell anemic patients with the
disease including: spleens 7 to 20 times enlarged (sometimes 10% of the
weight of the mouse), in vivo pathologies affecting the vital organs, and
congested livers. The tissues of the spleen, liver and kidney showed the
characteristic blockages associated with sickled cells. Similar to human
sickle cell patients, these blockages reduced blood flow and caused organ
deformities and damage.
(Ryan
et al., 1997)
Paszty
et al. developed the mouse line with both murine alpha -globin genes
deleted. This mouse line was bred with a line created by Ciavatta et al.
that lacked mbeta -globin to get a true heterozygotic line which was not
homologous for the mbeta-globin and m alpha -globin genes. This line was
then interbred with a sickle cell anemic mouse line developed by injecting
three different fragments of human DNA carrying the human alpha , beta
and human fetal hemoglobin genes G gamma and
A
gamma-globin along with the mini Locus Control Region (LCR
or the DNase I super-hypersensitive sites). Similar to Ryan et al., Paszty
et al. believed that the A gammaand G gamma globin genes would help the
transgenic mice to survive the gestation period due to their anti-sickling
properties. Paszty et al. saw approximately 4 to 26% expression of the
HbF at birth without continued expression to which they attribute the many
mouse deaths at birth.The figure below shows the low expression (in Part
B) of the human gamma-globin or human fetal globin and the resulting adult.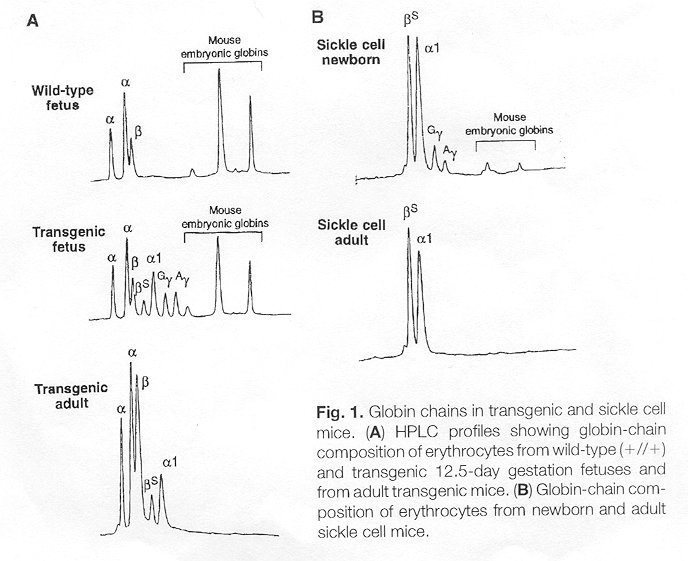
Their mice are slightly beta -thalassemic but have "normal
appearance, activity and fertility". They have found sickle cell anemia
symptoms in their mice such as irreversibly sickled cells (ISC), anemia,
increased rigidity of erythrocytes, and multiple organ damage. The ISC
formed in vitro from the HbF/HbS mice developed by Paszty et al. have the
classic sickle cell signs of decreased osmotic fragility, and increased
dynamic rigidity which are measured by osmotic gradient ektacytometry.
Multiple organ damage caused by the ISCs of the mice has also been characterized
using mouse organ histopathology. Damage to the sickle cell mice kidney,
lungs, heart, spleen and liver has been reported to be similar to damage
seen in the same organisms in sickle cell anemic patients. The heart and
kidney had twice the normal mass hypothetically due to increased function
and the spleen increased weight 13 times due to the accumulation of blocked
vessels and such related to ISCs. The mice lungs showed vascular congestion
and artifacts were found in the livers of the sickle cell anemic mice.
(Paszty
et al., 1997).
 Both
the Paszty et al. and the Ryan et al. transgenic mice show the complete
expression of only human hemoglobin. Though both are developed from the
same beta and alpha mouse hemoglobin mutants differing results were achieved
for the injected insertion of the human beta , alpha and gamma genes into
the mice embryos. While Paszty et al. say that they had "many" mice die
as a result of only 4 to 26% HbF globin expression at birth, Ryan et al.
observed levels of 30 to 50% HbF globin expression and approximately 90%
survival rate of the sickle cell anemic mice. These numbers clearly show
that while Paszty et al. has produced a transgenic mouse Ryan et al.s
mice are more viable. Paszty et al. had more analysis of the different
tissues damage rate, including heart and lung, and the osmolarity of the
sickle cells themselves. While these differences in data and results are
interesting the more important fact to consider is that both teams have
developed transgenic mice that can be ""indispensable"" (Barinaga,
1997).
Both
the Paszty et al. and the Ryan et al. transgenic mice show the complete
expression of only human hemoglobin. Though both are developed from the
same beta and alpha mouse hemoglobin mutants differing results were achieved
for the injected insertion of the human beta , alpha and gamma genes into
the mice embryos. While Paszty et al. say that they had "many" mice die
as a result of only 4 to 26% HbF globin expression at birth, Ryan et al.
observed levels of 30 to 50% HbF globin expression and approximately 90%
survival rate of the sickle cell anemic mice. These numbers clearly show
that while Paszty et al. has produced a transgenic mouse Ryan et al.s
mice are more viable. Paszty et al. had more analysis of the different
tissues damage rate, including heart and lung, and the osmolarity of the
sickle cells themselves. While these differences in data and results are
interesting the more important fact to consider is that both teams have
developed transgenic mice that can be ""indispensable"" (Barinaga,
1997).
<BACK
A PAGE
OUTLINE PAGE
To NEXT PAGE>